
6月1日に施行された令和6年度調剤報酬改定では、特定薬剤管理指導加算3が新設されました。
特定薬管理指導加算3はイとロにわかれ、イとロの算定要件はそれぞれさらに2つに分けられますが、イのうちの一方は以下のような算定要件になっています。
特定薬剤管理指導加算3のイ(一部抜粋)
RMPの策定が義務づけられている医薬品について、当該医薬品を新たに処方された場合に限り、患者又はその家族等に対し、RMPに基づきRMPに係る情報提供資材を活用し、副作用、併用禁忌等の当該医薬品の特性を踏まえ、適正使用や安全性等に関して十分な指導を行った場合
特定薬剤管理指導加算3については前回の記事でも解説しているので、そちらも参考にしてください!
今回の記事は特定薬剤管理指導加算3に登場する「RMP」について取り上げたいと思います。
もちろん特薬3のイについても深掘りしますが、今回の話の中心はRMP資材(RMPに係る情報提供資材)ではなく「RMP」についてです。
今回の改定で実感したのは、まだまだ RMPを知らない薬剤師が多いということです。
特薬3の登場によりRMP資材を知る人は増えるでしょうけど、RMP自体の理解は深まらないままなんて可能性も・・・。
特薬3を算定するしないに関わらず、RMPを活用することで服薬指導を充実させることが可能です。
RMPは患者さんに安心して薬を服用してもらえるようになるのはもちろん、薬剤師自身が安心して服薬指導を行うことを可能にしてくれるツールです。
ということで、今回は少しでも多くの方がRMPを理解して、活用できるようになってもらえることを目標として、RMP(医薬品リスク管理計画)について徹底解説!したいと思います。
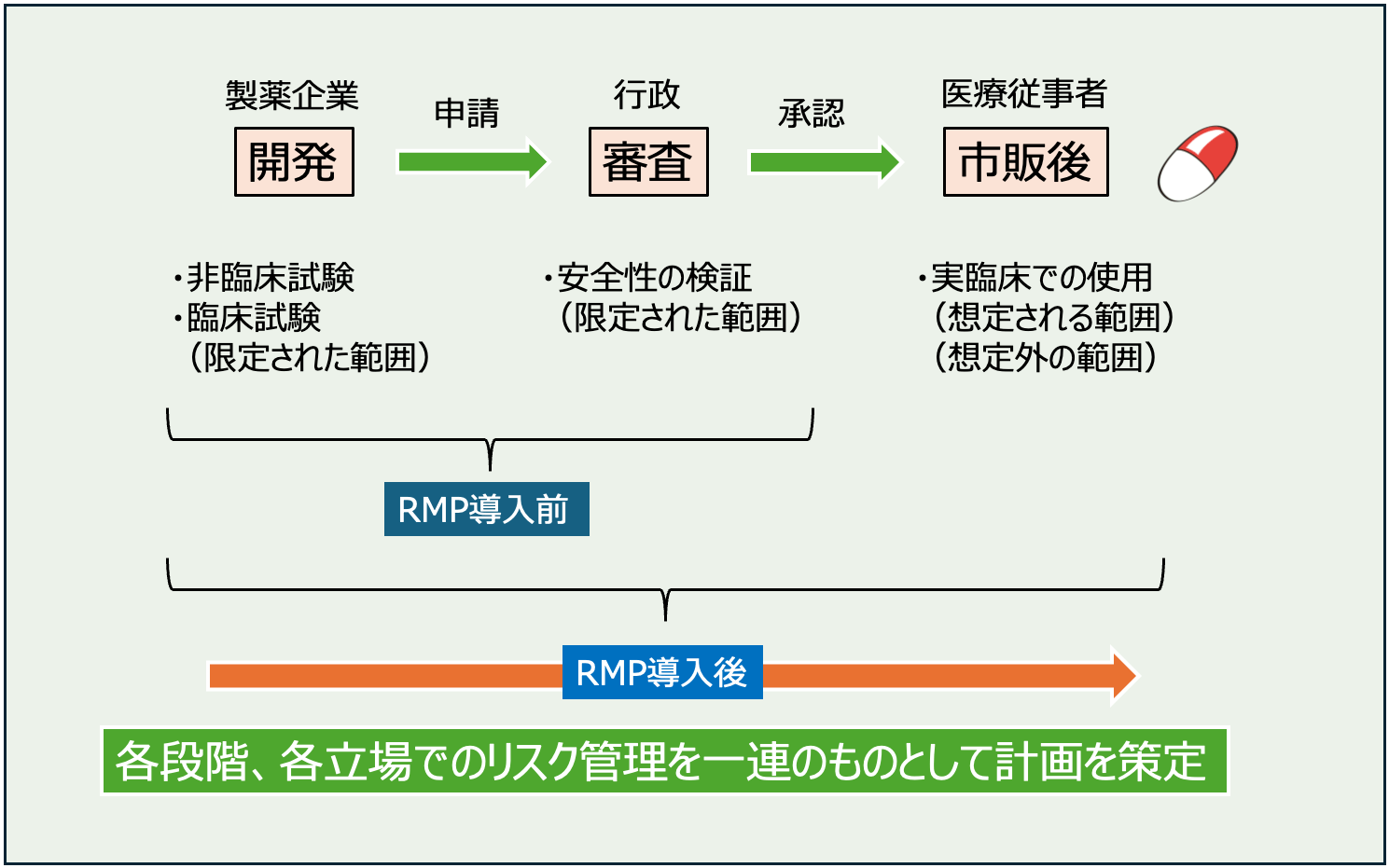
令和6年度調剤報酬改定で特定薬剤管理指導加算3が新設されたことに伴いRMPが注目されています。RMPとは医薬品のリスクについて、販売前の臨床試験等では確定できなかったものも含めて管理してくための計画です。開発・審査・市販後の各段階、各立場で考えられるリスクを一連として管理するための計画になります。RMPでは安全性検討事項をまとめ、医薬品安全監視活動とリスク最小化活動を計画・実施することにより、リスクを最小限に抑えていきます。リスク最小化活動の一つにRMP資材の作成・配布があり、患者向けRMP資材が作成されている場合、それを活用することが特定薬剤管理加算3のイの算定要件になっています。今回の改定により、患者向けRMP資材ばかりに注目が集まってしまいそうですが、加算の対象ではなくてもRMP自体や医療従事者向けRMP資材を使いこなすことで、患者さんに対する服薬指導を充実させることができ、新薬に対する薬剤師の不安感を払拭することが可能です。
1、そもそもRMPって何?
RMPとは医薬品リスク管理計画のことで、医薬品のリスクについて、開発・審査・市販後を通じて管理するための計画をまとめた資料になります。開発・審査の段階で明らかになっている副作用だけでは医薬品のリスクを確定するには不十分で、市販後に明らかになりうるものまでを想定して管理していくことが薬害を防ぐために重要です。
RMPとはRisk Management Planを略したもので、日本語で医薬品リスク管理計画と言います。
すべての医薬品にはリスク(副作用)がつきものです。
そのため、医薬品を開発する段階で非臨床試験や臨床試験が実施され、慎重に安全性のチェックが行われます。
ですが、市販後に使用される範囲に比べるとかなり限定的なものであることは否定できません。
開発段階で得られたデータは行政に提出され、有効性や安全性の評価に関する審査を受けます。
この段階で問題ないと判断されたものが承認され、医療用医薬品として販売されることになります。
市販開始後は開発段階よりも広い範囲で医薬品が使用されます。
高齢者や合併症のある患者、併用薬を服用中の患者など開発段階で十分に検証しきれていないようなケースに対して使用されていくことになります。
その結果、予期しないリスク(副作用)が発生したり、ベネフィット・リスクが損なわれる事象(副作用頻度の上昇、予想外の使用方法)が発生する可能性があります。
このように、医薬品の安全性を十分検証するためには、承認申請前の試験だけでは不十分で、市販後も継続して検証を行なっていくことが大切です。
そこで導入されたのが医薬品リスク管理計画(RMP)です。
RMPは開発〜審査の段階だけでなく、市販後まで含めた医薬品の安全性に関する計画です。
既知のものに限らず、未知のものまでを含めて、リスクを最小化するために実施される情報収集や情報提供についての計画がまとめられています。
日本では2012年4月に「医薬品リスク管理計画指針」が公開され、2013年4月に正式に導入されました。
導入以降は新薬の承認時にRMPの案を提出し、承認審査の段階でリスク最小化計画が必要と判断されたものについては、RMPの策定が義務付けられ、添付文書等とともにPMDAのホームページで公開されます。
その場合、RMP策定と実施が医薬品の承認条件となり、そのことが添付文書の21.承認条件の項に記載されています。

※添付文書への記載は改正GVP省令(2014年10月施行)で追加された内容のため、2014年9月末以前に申請した医薬品については未記載のものが存在します。
また、RMPの策定・実施が義務付けられている医薬品はPMDAのサイトにRMP提出品目一覧としてまとめられています。
1980年代から1990年代にかけて薬害肝炎問題(フィブリノゲン製剤や血液凝固第Ⅸ因子製剤を使用した患者がC型肝炎ウイルスに感染してしまった事件)が発生しました。国はその再発防止のために委員会を立ち上げ、検証を重ね、2010年に「薬害肝炎検証・検討委員会「最終提言」について」が公表しました。その中で、欧米の制度を参考に「リスク最小化計画実施制度」を導入し、ICH-E2Eガイドライン(医薬品規制調和国際会議の安全性監視活動計画ガイドライン)に沿って「医薬品安全性監視の方法」を取り入れた「医薬品リスク管理」を適切に実施することが求められ、その結果、導入されることになったのがRMP(医薬品リスク管理計画)です。
2、RMPに記載されている情報
RMPには安全性検討事項として重要な特定されたリスク、重要な潜在的リスク、重要な不足情報がまとめられています。また、それらのリスクを管理するために、医薬品安全性監視活動として追加の情報収集活動が実施され、さらにリスク最小化活動として追加の情報提供活動が実施されます。RMPの策定が義務付けられている医薬品はこれらの計画について必要に応じて見直しが行われます。
特定の医薬品のRMPを読みたい場合はPMDAの医療用医薬品 添付文書等情報検索で確認することができます。
また、製薬企業の医療従事者向けページに掲載されている個別の製品情報からも閲覧可能です。
まずは RMPの「R」の部分、リスクが指す内容について解説します。
RMPで扱うリスクは以下の3つに分類されており、これらを合わせて「安全性検討事項」と呼びます。
